Sequence Databases¶
The NCBI Sequence Database¶
All published genome sequences are available over the internet, as it is a requirement of every scientific journal that any published DNA or RNA or protein sequence must be deposited in a public database. The main resources for storing and distributing sequence data are three large databases: the NCBI database (www.ncbi.nlm.nih.gov/), the European Molecular Biology Laboratory (EMBL) database (www.ebi.ac.uk/embl/, and the DNA Database of Japan (DDBJ) database (www.ddbj.nig.ac.jp/). These databases collect all publicly available DNA, RNA and protein sequence data and make it available for free. They exchange data nightly, so contain essentially the same data.
In this chapter we will discuss the NCBI database. Note however that it contains essentially the same data as in the EMBL/DDBJ databases.
Sequences in the NCBI Sequence Database (or EMBL/DDBJ) are identified by an accession number. This is a unique number that is only associated with one sequence. For example, the accession number NC_001477 is for the DEN-1 Dengue virus genome sequence. The accession number is what identifies the sequence. It is reported in scientific papers describing that sequence.
As well as the sequence itself, for each sequence the NCBI database (or EMBL/DDBJ databases) also stores some additional annotation data, such as the name of the species it comes from, references to publications describing that sequence, etc. Some of this annotation data was added by the person who sequenced a sequence and submitted it to the NCBI database, while some may have been added later by a human curator working for NCBI.
The NCBI database contains several sub-databases, the most important of which are:
- the NCBI Nucleotide database: contains DNA and RNA sequences
- the NCBI Protein database: contains protein sequences
- EST: contains ESTs (expressed sequence tags), which are short sequences derived from mRNAs
- the NCBI Genome database: contains DNA sequences for whole genomes
- PubMed: contains data on scientific publications
Searching for an accession number in the NCBI database¶
In the DNA Sequence Statistics chapter (1), you learnt how to obtain a FASTA file containing the DNA sequence corresponding to a particular accession number, eg. accession number NC_001477 (the DEN-1 Dengue virus genome sequence), either via the NCBI website or using the getncbiseq() function in R.
As explained in the DNA Sequence Statistics (1) chapter, the FASTA format is a file format commonly used to store sequence information. The first line starts with the character ‘>’ followed by a name and/or description for the sequence. Subsequent lines contain the sequence itself.
>mysequence1
ACATGAGACAGACAGACCCCCAGAGACAGACCCCTAGACACAGAGAGAG
TATGCAGGACAGGGTTTTTGCCCAGGGTGGCAGTATG
A FASTA file can contain more than one sequence. If a FASTA file contains many sequences, then for each sequence it will have a header line starting with ‘>’ followed by the sequence itself.
>mysequence1
ACATGAGACAGACAGACCCCCAGAGACAGACCCCTAGACACAGAGAGAG
TATGCAGGACAGGGTTTTTGCCCAGGGTGGCAGTATG
>mysequence2
AGGATTGAGGTATGGGTATGTTCCCGATTGAGTAGCCAGTATGAGCCAG
AGTTTTTTACAAGTATTTTTCCCAGTAGCCAGAGAGAGAGTCACCCAGT
ACAGAGAGC
NCBI Sequence Format (NCBI Format)¶
As mentioned above, for each sequence the NCBI database stores some extra information such as the species that it came from, publications describing the sequence, etc. This information is stored in the NCBI entry or NCBI record for the sequence. The NCBI entry for a sequence can be viewed by searching the NCBI database for the accession number for that sequence. The NCBI entries for sequences are stored in a particular format, known as NCBI format.
To view the NCBI entry for the DEN-1 Dengue virus (which has accession NC_001477), follow these steps:
- Go to the NCBI website (www.ncbi.nlm.nih.gov).
- Search for the accession number.
- On the results page, if your sequence corresponds to a nucleotide (DNA or RNA) sequence, you should see a hit in the Nucleotide database, and you should click on the word ‘Nucleotide’ to view the NCBI entry for the hit. Likewise, if your sequence corresponds to a protein sequence, you should see a hit in the Protein database, and you should click on the word ‘Protein’ to view the NCBI entry for the hit.
- After you click on ‘Nucleotide’ or ‘Protein’ in the previous step, the NCBI entry for the accession will appear.
For example, the NCBI entry for the DEN-1 Dengue virus genome sequence (NCBI accession NC_001477) looks like this:
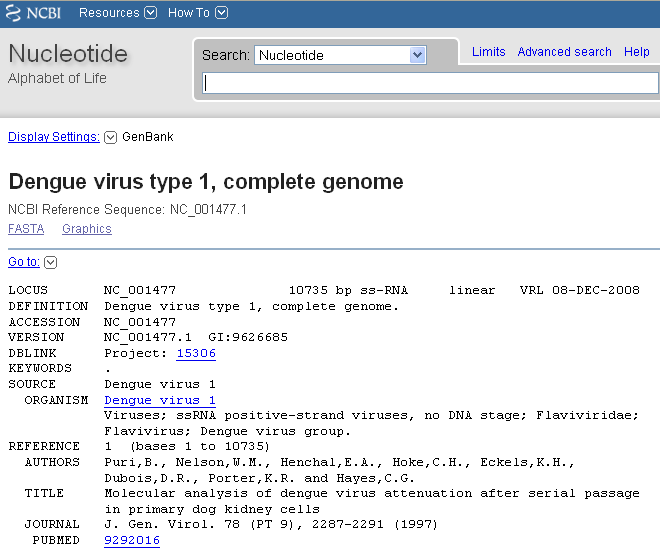
The NCBI entry for an accession contains a lot of information about the sequence, such as papers describing it, features in the sequence, etc. The ‘DEFINITION’ field gives a short description for the sequence. The ‘ORGANISM’ field in the NCBI entry identifies the species that the sequence came from. The ‘REFERENCE’ field contains scientific publications describing the sequence. The ‘FEATURES’ field contains information about the location of features of interest inside the sequence, such as regulatory sequences or genes that lie inside the sequence. The ‘ORIGIN’ field gives the sequence itself.
RefSeq¶
When carrying out searches of the NCBI database, it is important to bear in mind that the database may contain redundant sequences for the same gene that were sequenced by different laboratories (because many different labs have sequenced the gene, and submitted their sequences to the NCBI database).
There are also many different types of nucleotide sequences and protein sequences in the NCBI database. With respect to nucleotide sequences, some many be entire genomic DNA sequences, some may be mRNAs, and some may be lower quality sequences such as expressed sequence tags (ESTs, which are derived from parts of mRNAs), or DNA sequences of contigs from genome projects.
Furthermore, some sequences may be manually curated so that the associated entries contain extra information, but the majority of sequences are uncurated.
As mentioned above, the NCBI database often contains redundant information for a gene, contains sequences of varying quality, and contains both uncurated and curated data.
As a result, NCBI has made a special database called RefSeq (reference sequence database), which is a subset of the NCBI database. The data in RefSeq is manually curated, is high quality sequence data, and is non-redundant; this means that each gene (or splice-form of a gene, in the case of eukaryotes), protein, or genome sequence is only represented once.
The data in RefSeq is curated and is of much higher quality than the rest of the NCBI Sequence Database. However, unfortunately, because of the high level of manual curation required, RefSeq does not cover all species, and is not comprehensive for the species that are covered so far.
You can easily tell that a sequence comes from RefSeq because its accession number starts with particular sequence of letters. That is, accessions of RefSeq sequences corresponding to protein records usually start with ‘NP_’, and accessions of RefSeq curated complete genome sequences usually start with ‘NC_’ or ‘NS_’.
Querying the NCBI Database¶
You may need to interrogate the NCBI Database to find particular sequences or a set of sequences matching given criteria, such as:
- The sequence with accession NC_001477
- The sequences published in Nature 460:352-358
- All sequences from Chlamydia trachomatis
- Sequences submitted by Matthew Berriman
- Flagellin or fibrinogen sequences
- The glutamine synthetase gene from Mycobacteriuma leprae
- The upstream control region of the Mycobacterium leprae dnaA gene
- The sequence of the Mycobacterium leprae DnaA protein
- The genome sequence of Trypanosoma cruzi
- All human nucleotide sequences associated with malaria
There are two main ways that you can query the NCBI database to find these sets of sequences. The first possibility is to carry out searches on the NCBI website. The second possiblity is to carry out searches from R.
Below, we will explain how to use both methods to carry out queries on the NCBI database. In general, the two methods should give the same result, but in some cases they do not, for various reasons, as shall be explained below.
Querying the NCBI Database via the NCBI Website¶
If you are carrying out searches on the NCBI website, to narrow down your searches to specific types of sequences or to specific organisms, you will need to use “search tags”.
For example, the search tags “[PROP]” and “[ORGN]” let you restrict your search to a specific subset of the NCBI Sequence Database, or to sequences from a particular taxon, respectively. Here is a list of useful search tags, which we will explain how to use below:
| Search tag | Example | Restricts your search to sequences: |
|---|---|---|
| [AC] | NC_001477[AC] | With a particular accession number |
| [ORGN] | Fungi[ORGN] | From a particular organism or taxon |
| [PROP] | biomol_mRNA[PROP] | Of a specific type (eg. mRNA) or from a specific database (eg. RefSeq) |
| [JOUR] | Nature[JOUR] | Described in a paper published in a particular journal |
| [VOL] | 531[VOL] | Described in a paper published in a particular journal volume |
| [PAGE] | 27[PAGE] | Described in a paper with a particular start-page in a journal |
| [AU] | “Smith J”[AU] | Described in a paper, or submitted to NCBI, by a particular author |
To carry out searches of the NCBI database, you first need to go to the NCBI website, and type your search query into the search box at the top. For example, to search for all sequences from Fungi, you would type “Fungi[ORGN]” into the search box on the NCBI website.
You can combine the search tags above by using “AND”, to make more complex searches. For example, to find all mRNA sequences from Fungi, you could type “Fungi[ORGN] AND biomol_mRNA[PROP]” in the search box on the NCBI website.
Likewise, you can also combine search tags by using “OR”, for example, to search for all mRNA sequences from Fungi or Bacteria, you would type “(Fungi[ORGN] OR Bacteria[ORGN]) AND biomol_mRNA[PROP]” in the search box. Note that you need to put brackets around “Fungi[ORGN] OR Bacteria[ORGN]” to specify that the word “OR” refers to these two search tags.
Here are some examples of searches, some of them made by combining search terms using “AND”:
| Typed in the search box | Searches for sequences: |
|---|---|
| NC_001477[AC] | With accession number NC_001477 |
| Nature[JOUR] AND 460[VOL] AND 352[PAGE] | Published in Nature 460:352-358 |
| “Chlamydia trachomatis”[ORGN] | From the bacterium Chlamydia trachomatis |
| “Berriman M”[AU] | Published in a paper, or submitted to NCBI, by M. Berriman |
| flagellin OR fibrinogen | Which contain the word ‘flagellin’ or ‘fibrinogen’ in their NCBI record |
| “Mycobacterium leprae”[ORGN] AND dnaA | Which are from M. leprae, and contain “dnaA” in their NCBI record |
| “Homo sapiens”[ORGN] AND “colon cancer” | Which are from human, and contain “colon cancer” in their NCBI record |
| “Homo sapiens”[ORGN] AND malaria | Which are from human, and contain “malaria” in their NCBI record |
| “Homo sapiens”[ORGN] AND biomol_mrna[PROP] | Which are mRNA sequences from human |
| “Bacteria”[ORGN] AND srcdb_refseq[PROP] | Which are RefSeq sequences from Bacteria |
| “colon cancer” AND srcdb_refseq[PROP] | From RefSeq, which contain “colon cancer” in their NCBI record |
Note that if you are searching for a phrase such as “colon cancer” or “Chlamydia trachomatis, you need to put the phrase in inverted commas when typing it into the search box. This is because if you type the phrase in the search box without using inverted commas, the search will be for NCBI records that contain either of the two words ‘colon’ or ‘cancer’ (or either of the two words ‘Chlamydia’ or ‘trachomatis’), not necessarily both words.
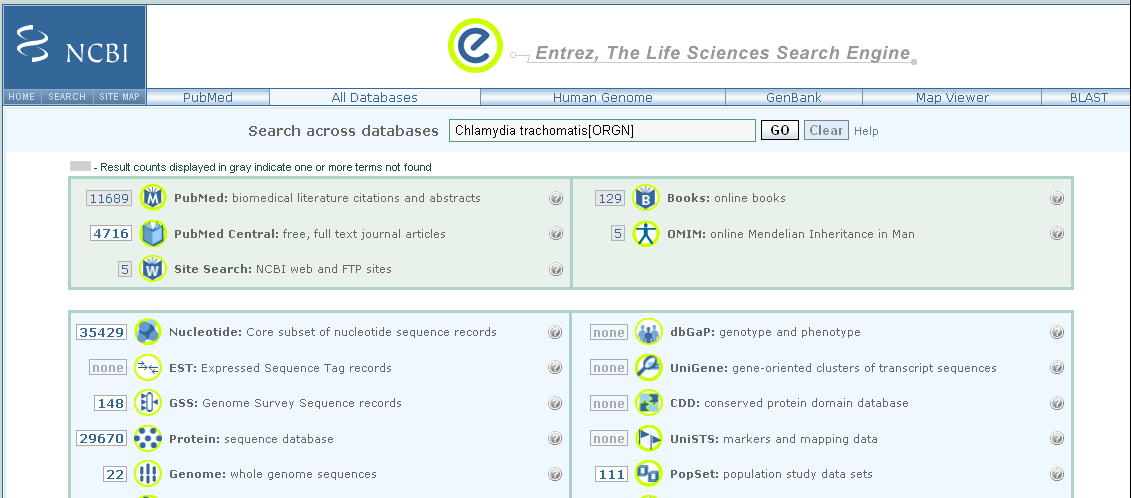
As mentioned above, the NCBI database contains several sub-databases, including the NCBI Nucleotide database and the NCBI Protein database. If you go to the NCBI website, and type one of the search queries above in the search box at the top of the page, the results page will tell you how many matching NCBI records were found in each of the NCBI sub-databases.
For example, if you search for “Chlamydia trachomatis[ORGN]”, you will get matches to proteins from C. trachomatis in the NCBI Protein database, matches to DNA and RNA sequences from C. trachomatis in the NCBI Nucleotide database, matches to whole genome sequences for C. trachomatis strains in the NCBI Genome database, and so on:
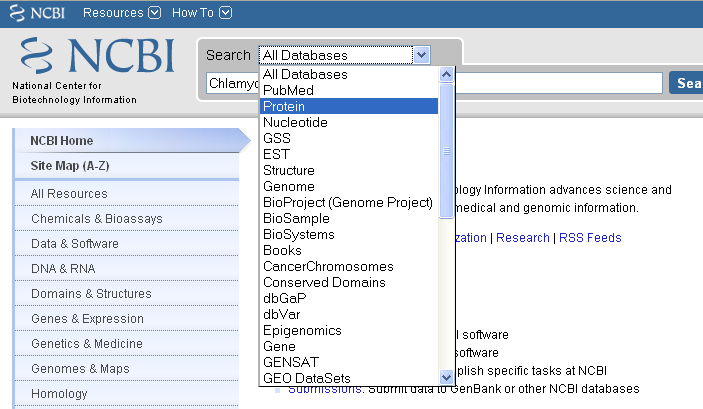
Alternatively, if you know in advance that you want to search a particular sub-database, for example, the NCBI Protein database, when you go to the NCBI website, you can select that sub-database from the drop-down list above the search box, so that you will search that sub-database:
Example: finding the sequences published in Nature 460:352-358¶
For example, if you want to find sequences published in Nature 460:352-358, you can use the “[JOUR]”, “[VOL]” and “[PAGE]” search terms. That is, you would go to the NCBI website and type in the search box on the top: “Nature”[JOUR] AND 460[VOL] AND 352[PAGE], where [JOUR] specifies the journal name, [VOL] the volume of the journal the paper is in, and [PAGE] the page number.

This should bring up a results page with “50890” beside the word “Nucleotide”, and “1” beside the word “Genome”, and “25701” beside the word “Protein”, indicating that there were 50890 hits to sequence records in the Nucleotide database, which contains DNA and RNA sequences, and 1 hit to the Genome database, which contains genome sequences, and 25701 hits to the Protein database, which contains protein sequences:

If you click on the word “Nucleotide”, it will bring up a webpage with a list of links to the NCBI sequence records for those 50890 hits. The 50890 hits are all contigs from the schistosome worm Schistosoma mansoni.
Likewise, if you click on the word “Protein”, it will bring up a webpage with a list of links to the NCBI sequence records for the 25701 hits, and you will see that the hits are all predicted proteins for Schistosoma mansoni.
If you click on the word “Genome”, it will bring you to the NCBI record for the Schistosoma mansoni genome sequence, which has NCBI accession NS_00200. Note that the accession starts with “NS_”, which indicates that it is a RefSeq accession.
Therefore, in Nature volume 460, page 352, the Schistosoma mansoni genome sequence was published, along with all the DNA sequence contigs that were sequenced for the genome project, and all the predicted proteins for the gene predictions made in the genome sequence. You can view the original paper on the Nature website at http://www.nature.com/nature/journal/v460/n7253/abs/nature08160.html.
Note: Schistmosoma mansoni is a parasitic worm that is responsible for causing schistosomiasis, which is classified by the WHO as a neglected tropical disease.
Querying the NCBI Database via R¶
Instead of carrying out searches of the NCBI database on the NCBI website, you can carry out searches directly from R by using the SeqinR R package.
It is possible to use the SeqinR R package to retrieve sequences from these databases. The SeqinR package was written by the group that created the ACNUC database in Lyon, France (http://pbil.univ-lyon1.fr/databases/acnuc/acnuc.html). The ACNUC database is a database that contains most of the data from the NCBI Sequence Database, as well as data from other sequence databases such as UniProt and Ensembl.
An advantage of the ACNUC database is that it brings together data from various different sources, and makes it easy to search, for example, by using the SeqinR R package.
As will be explained below, the ACNUC database is organised into various different ACNUC (sub)-databases, which contain different parts of the NCBI database, and when you want to search the NCBI database via R, you will need to specify which ACNUC sub-database the NCBI data that you want to query is stored in.
To obtain a full list of the ACNUC sub-databases that you can access using SeqinR, you can use the “choosebank()” function from SeqinR:
> library("seqinr") # Load the SeqinR R package
> choosebank() # List all the sub-databases in ACNUC
[1] "genbank" "embl" "emblwgs" "swissprot"
[5] "ensembl" "hogenom" "hogenomdna" "hovergendna"
[9] "hovergen" "hogenom4" "hogenom4dna" "homolens"
[13] "homolensdna" "hobacnucl" "hobacprot" "phever2"
[17] "phever2dna" "refseq" "nrsub" "greviews"
[21] "bacterial" "protozoan" "ensbacteria" "ensprotists"
[25] "ensfungi" "ensmetazoa" "ensplants" "mito"
[29] "polymorphix" "emglib" "taxobacgen" "refseqViruses"
Alas, the ACNUC sub-databases do not have a one-to-one correspondence with the NCBI sub-databases (the NCBI Protein database, NCBI EST database, NCBI Genome database, etc.)!
Three of the most important sub-databases in ACNUC which can be searched from R are:
- “genbank”: this contains DNA and RNA sequences from the NCBI Sequence Database, except for certain classes of sequences (eg. draft genome sequence data from genome sequencing projects)
- “refseq”: this contains DNA and RNA sequences from Refseq, the curated part of the NCBI Sequence Database
- “refseqViruses”: this contains DNA, RNA and proteins sequences from viruses from RefSeq
You can find more information about what each of these ACNUC databases contains by looking at the ACNUC website.
You can carry out complex queries using the “query()” function from the SeqinR package. If you look at the help page for the query() function (by typing “help(query)”, you will see that it allows you to specify criteria that you require the sequences to fulfill.
For example, to search for a sequence with a particular NCBI accession, you can use the “AC=” argument in “query()”. The “query()” function will then search for sequences in the NCBI Sequence Database that match your criteria.
Just as you can use “AC=” to specify an accession in a search, you can specify that you want to find sequences whose NCBI records contain a certain keywords by using “K=” as an argument (input) to the “query()” function. Likewise you can limit a search to either DNA or mRNA sequences by using the “M=” argument for the “query()” function. Here are some more possible arguments you can use in the “query()” function:
| Argument | Example | Restricts your search to sequences: |
|---|---|---|
| “AC=” | “AC=NC_001477” | With a particular accession number |
| “SP=” | “SP=Chlamydia” | From a particular organism or taxon |
| “M=” | “M=mRNA” | Of a specific type (eg. mRNA) |
| “J=” | “J=Nature” | Described in a paper published in a particular journal |
| “R=” | “R=Nature/460/352” | Described in a paper in a particular journal, volume and start-page |
| “AU=” | “AU=Smith” | Described in a paper, or submitted to NCBI, by a particular author |
The full list of possible arguments for the “query()” funtion are given on its help page. Here are some examples using the query function:
| Input to the query() function | Searches for sequences: |
|---|---|
| “AC=NC_001477” | With accession number NC_001477 |
| “R=Nature/460/352” | Published in Nature 460:352-358 |
| “SP=Chlamydia trachomatis” | From the bacterium Chlamydia trachomatis |
| “AU=Berriman” | Published in a paper, or submitted to NCBI, by someone called Berriman |
| “K=flagellin OR K=fibrinogen” | Which have the keyword ‘flagellin’ or ‘fibrinogen’ |
| “SP=Mycobacterium leprae AND K=dnaA” | Which are from M. leprae, and have the keyword “dnaA” |
| “SP=Homo sapiens AND K=colon cancer” | Which are from human, and have the keyword “colon cancer” |
| “SP=Homo sapiens AND K=malaria” | Which are from human, and have the keyword “malaria” |
| “SP=Homo sapiens AND M=mrna” | Which are mRNA sequences from human |
| “SP=Bacteria” | Which are sequences from Bacteria |
As explained above, the ACNUC database contains the NCBI sequence data organised into several sub-databases, and you can view the list of those sub-databases by using the “choosebank()” function from the SeqinR package. When you want to use “query()” to carry out a particular sub-database (eg. “genbank”, which contains DNA and RNA sequences from the NCBI Sequence Database), you need to first specify the database that you want to search by using the “choosebank()” function, for example:
> choosebank("genbank") # Specify that we want to search the 'genbank' ACNUC sub-database
Likewise, to specify that we want to search the ‘refseq’ ACNUC sub-database, which contains sequences from the NCBI RefSeq database, we would type:
> choosebank("refseq") # Specify that we want to search the 'refseq' ACNUC sub-database
Once you have specified which ACNUC sub-database you want to search, you can carry out a search of that sub-database by using the “query()” function. You need to pass the “query()” function both a name for your query (which you can make up), and the query itself (which will be in the format of the examples in the table above). For example, if we want to search for RefSeq sequences from Bacteria, we might decide to call our query “RefSeqBact”, and we would call the “query()” function as follows:
> query("RefSeqBact", "SP=Bacteria")
As explained below, the results of the search are stored in a list variable called “RefSeqBact”, and can be retrieved from that list variable. The last thing to do once you have completed your search is to close the connection to the ACNUC sub-database that you were searching, by typing:
> closebank()
Thus, there are three steps involved in carrying out a query using SeqinR: first use “choosebank()” to select the ACNUC sub-database to search, secondly use “query()” to query the database, and thirdly use “closebank()” to close the connection to the ACNUC sub-database.
Another example could be to search for mRNA sequences from the parasitic worm Schistosoma mansoni in the NCBI Nucleotide database. The appropriate ACNUC sub-database to search is the “genbank” ACNUC sub-database. We may decide to call our search “SchistosomamRNA”. Therefore, to carry out the search, we type in R:
> choosebank("genbank")
> query("SchistosomamRNA", "SP=Schistosoma mansoni AND M=mrna")
> closebank()
Example: finding the sequence for the DEN-1 Dengue virus genome¶
Another example could be to search for the DEN-1 Dengue virus genome sequence, which has accession NC_001477. This is a viral genome sequence, and so should be in the ACNUC sub-database “refSeqViruses”. Thus to search for this sequence, calling our search “Dengue1”, we type in R:
> choosebank("refseqViruses")
> query("Dengue1", "AC=NC_001477")
The result of the search is now stored in the list variable Dengue1. Remember that a list is an R object that is like a vector, but can contain elements that are numeric and/or contain characters. In this case, the list Dengue1 contains information on the NCBI records that match the query (ie. information on the NCBI record for accession NC_001477).
If you look at the help page for “query()”, the details of the arguments are given under the heading “Arguments”, and the details of the results (outputs) are given under the heading “Value”. If you read this now, you will see that it tells us that the result of the “query()” function is a list with six different named elements, named “call”, “name”, “nelem”, “typelist”, “req”, and “socket”. The content of each of these six named elements is explained, for example, the “nelem” element contains the number of sequences that match the query, and the “req” element contains their accession numbers.
In our example, the list object Dengue1 is an output of the “query()” function, and so has each of these six named elements, as we can find out by using the “attributes()” function, and looking at the named elements listed under the heading “$names”:
> attributes(Dengue1)
$names
[1] "call" "name" "nelem" "typelist" "req" "socket"
$class
[1] "qaw"
As explained in the brief introduction to R, we can retrieve the value for each of the named elements in the list Dengue1 by using “$”, followed by the element’s name, for example, to get the value of the element named “nelem” in the list Dengue1, we type:
> Dengue1$nelem
[1] 1
This tells us that there was one sequence in the ‘refseqViruses’ ACNUC database that matched the query. This is what we would expect, as there should only be one sequence corresponding to accession NC_001477.
To obtain the accession numbers of the sequence found, we can type:
> Dengue1$req
[[1]]
name length frame ncbicg
"NC_001477" "10735" "0" "1"
As expected, the accession number of the matching sequence is NC_001477.
When you type “attributes(Dengue1)” you can see that there are two headings, “$names”, and “$class”. As explained above, the named elements of the list variable Dengue1 are listed under the heading “$names”. In fact, the headings “$names” and “$class” are two attributes of the list variable Dengue1. We can retrieve the values of the attributes of a variable using the “attr()” function. For example, to retrieve the value of the attribute “$names” of Dengue1, we type:
> attr(Dengue1, "names")
[1] "call" "name" "nelem" "typelist" "req" "socket"
This gives us the value of the attribute “$names”, which contains the the names of the named elements of the list variable Dengue1. Similarly, we can retrieve the value of the a attribute “$class” of Dengue1, we type:
> attr(Dengue1, "class")
[1] "qaw"
This tells us that the value of the attribute “$class” is “qaw”.
The final step in retrieving a genomic DNA sequence is to use the “getSequence()” function to tell R to retrieve the sequence data. The command below uses “getSequence()” to retrieve the sequence data for the DEN-1 Dengue virus genome, and puts the sequence into a variable dengueseq:
> dengueseq <- getSequence(Dengue1$req[[1]])
Note that the input to the getSequence() command is Dengue1$req[[1]], which contains the name of the NCBI record that the list Dengue1 contains information about.
Once you have retrieved a sequence, you can then print it out. The variable dengueseq is a vector containing the nucleotide sequence. Each element of the vector contains one nucleotide of the sequence. Therefore, we can print out the first 50 nucleotides of the DEN-1 Dengue genome sequence by typing:
> dengueseq[1:50]
[1] "a" "g" "t" "t" "g" "t" "t" "a" "g" "t" "c" "t" "a" "c" "g" "t" "g" "g" "a"
[20] "c" "c" "g" "a" "c" "a" "a" "g" "a" "a" "c" "a" "g" "t" "t" "t" "c" "g" "a"
[39] "a" "t" "c" "g" "g" "a" "a" "g" "c" "t" "t" "g"
Note that dengueseq[1:50] refers to the elements of the vector dengueseq with indices from 1-50. These elements contain the first 50 nucleotides of the DEN-1 Dengue virus genome sequence.
As well as retrieving the DNA (or RNA or protein) sequence itself, SeqinR can also retrieve all the annotations for the sequence, for example, information on when the sequence was sequenced, who sequenced it, what organism is it from, what paper was it described in, what genes were identified in the sequence, and so on.
Once you have retrieved a sequence using SeqinR, you can retrieved its annotations by using the “getAnnot()” function. For example, to view the annotations for the DEN-1 Dengue virus genome sequence, we type:
> annots <- getAnnot(Dengue1$req[[1]])
This stores the annotations information from the NCBI record for the DEN-1 Dengue virus sequence in a vector variable annots, with one line of the NCBI record in each element of the vector. Therefore, we can print out the first 20 lines of the NCBI record by typing:
> annots[1:20]
[1] "LOCUS NC_001477 10735 bp ss-RNA linear VRL 08-DEC-2008"
[2] "DEFINITION Dengue virus type 1, complete genome."
[3] "ACCESSION NC_001477"
[4] "VERSION NC_001477.1 GI:9626685"
[5] "DBLINK Project: 15306"
[6] "KEYWORDS ."
[7] "SOURCE Dengue virus 1"
[8] " ORGANISM Dengue virus 1"
[9] " Viruses; ssRNA positive-strand viruses, no DNA stage; Flaviviridae;"
[10] " Flavivirus; Dengue virus group."
[11] "REFERENCE 1 (bases 1 to 10735)"
[12] " AUTHORS Puri,B., Nelson,W.M., Henchal,E.A., Hoke,C.H., Eckels,K.H.,"
[13] " Dubois,D.R., Porter,K.R. and Hayes,C.G."
[14] " TITLE Molecular analysis of dengue virus attenuation after serial passage"
[15] " in primary dog kidney cells"
[16] " JOURNAL J. Gen. Virol. 78 (PT 9), 2287-2291 (1997)"
[17] " PUBMED 9292016"
[18] "REFERENCE 2 (bases 1 to 10735)"
[19] " AUTHORS McKee,K.T. Jr., Bancroft,W.H., Eckels,K.H., Redfield,R.R.,"
[20] " Summers,P.L. and Russell,P.K."
On the left of the annotations, you will see that there is a column containing the field name. For example, the line of the with “ACCESSION” in the left column is the accession field, which contains the accession for the sequence (NC_001477 for the DEN-1 Dengue virus).
The line with “ORGANISM” in the left column is the organism field, and usually contains the Latin name for the organism (“Dengue virus 1” here). The line with “AUTHORS” in the left column is the authors field, and contain the names of authors that wrote papers to describe the sequence and/or the names of the people who submitted the sequence to the NCBI Database.
When you have finished your running your query and getting the corresponding sequences and annotations, close the connection to the ACNUC sub-database:
> closebank()
Example: finding the sequences published in Nature 460:352-358¶
We described above how to search for the sequences published in Nature 460:352-358, using the NCBI website. A second method is to use the SeqinR R package to search the ACNUC databases (which contain the NCBI sequence data) from R.
If you look at the help page the “query()” function, you see that you can query for sequences published in a particular paper using R=refcode, specifying the reference as refcode such as in jcode/volume/page (e.g., JMB/13/5432 or R=Nature/396/133). For the paper Nature 460:352-358, we would need to use the refcode ‘R=Nature/460/352’.
First we need to specify which of the ACNUC databases we want to search. For example, to specify that we want to search the “genbank” ACNUC database, which contains DNA and RNA sequences from the NCBI Nucleotide database, we type:
> choosebank("genbank") # Specify that we want to search the 'genbank' ACNUC sub-database
We can then search the ‘genbank’ database for sequences that match a specific set of criteria by using the “query()” function. For example, to search for sequences that were published in Nature 460:352-358, we type:
> query('naturepaper', 'R=Nature/460/352')
The line above tells R that we want to store the results of the query in an R list variable called naturepaper. To get the value of the element named “nelem” in the list naturepaper, we type:
> naturepaper$nelem
[1] 19022
This tells us that there were 19022 sequences in the ‘genbank’ ACNUC database that matched the query. The ‘genbank’ ACNUC database contains DNA or RNA sequences from the NCBI Nucleotide database. Why don’t we get the same number of sequences as found by carrying out the search on the NCBI website (where we found 50890 hits to the NCBI Nucleotide database)? The reason is that the ACNUC ‘genbank’ database does not contain all the sequences in the NCBI Nucleotide database, for example, it does not contain sequences that are in RefSeq or many short DNA sequences from sequencing projects.
To obtain the accession numbers of the first five of the 19022 sequences, we can type:
> accessions <- naturepaper$req
> accessions[1:5]
[[1]]
name length frame ncbicg
"FN357292" "4179495" "0" "1"
[[2]]
name length frame ncbicg
"FN357293" "2211188" "0" "1"
[[3]]
name length frame ncbicg
"FN357294" "1818661" "0" "1"
[[4]]
name length frame ncbicg
"FN357295" "2218116" "0" "1"
[[5]]
name length frame ncbicg
"FN357296" "3831198" "0" "1"
This tells us that the NCBI accessions of the first five sequences (of the 19022 DNA or RNA sequences found that were published in Nature 460:352-358) are FN357292, FN357293, FN357294, FN357295, and FN357296.
To retrieve these first five sequences, and print out the first 10 nucleotide bases of each sequence, we use the getSequence() command, typing:
> for (i in 1:5)
{
seqi <- getSequence(naturepaper$req[[i]])
print(seqi[1:10])
}
[1] "t" "t" "g" "t" "c" "g" "a" "t" "t" "a"
[1] "g" "g" "t" "c" "c" "t" "t" "a" "a" "g"
[1] "g" "c" "c" "t" "g" "a" "c" "c" "a" "t"
[1] "t" "a" "t" "t" "t" "c" "c" "a" "a" "t"
[1] "c" "a" "a" "t" "c" "a" "c" "t" "c" "a"
Note that the input to the getSequence() command is Dengue1$req[[i]], which contains the name of i th NCBI record that the list naturepaper contains information about.
Once we have carried out our queries and retrieved the sequences, the final step is to close the connection to the ACNUC sub-database that we searched (“genbank” here):
> closebank()
Saving sequence data in a FASTA-format file¶
Once you have retrieved a sequence, or set of sequences from the NCBI Database, using SeqinR, it is conveninent to save the sequences in a file in FASTA format. This can be done using the “write.fasta()” function in the SeqinR package, which was introduced in Chapter 1.
If you look at the help page for the “write.fasta()” function, you will see that as input it takes a list of vectors, where each vector contains one DNA, RNA or protein sequence.
For example, if you retrieve the sequences of human tRNAs from the NCBI Database by querying the ACNUC “genbank” sub-database, you can save the sequences in a FASTA format file called “humantRNAs.fasta” by typing:
> choosebank("genbank") # select the ACNUC sub-database to be searched
> query("humtRNAs", "SP=homo sapiens AND M=TRNA") # specify the query
> myseqs <- getSequence(humtRNAs) # get the sequences
> mynames <- getName(humtRNAs) # get the names of the sequences
> write.fasta(myseqs, mynames, file.out="humantRNAs.fasta")
> closebank()
In the above code, we get the sequences of the human tRNAs using the function “getSequence()” from the SeqinR package. We also use a function “getName()” from the SeqinR package to get the sequences’ names. Then we use the “write.fasta()” function to write the sequences to a FASTA file “humantRNAs.fasta”. The “write.fasta()” takes as arguments: the list myseqs containing the sequences, the list mynames containing the names of the sequences, and the name of the output file (“humantRNAs.fasta” here).
Finding the genome sequence for a particular species¶
Microbial genomes are generally smaller than eukaryotic genomes (Escherichia coli has about 5 million base pair in its genome, while the human genome is about 3 billion base pairs). Because they are considerably less expensive to sequence, many microbial genome sequencing projects have been completed.
If you don’t know the accession number for a genome sequence (eg. for Mycobacterium leprae, the bacterium that causes leprosy), how can you find it out?
The easiest way to do this is to look at the NCBI Genome website, which lists all fully sequenced genomes and gives the accession numbers for the corresponding DNA sequences.
If you didn’t know the accession number for the Mycobacterium leprae genome, you could find it on the NCBI Genome website by following these steps:
- Go to the NCBI Genome website (http://www.ncbi.nlm.nih.gov/sites/entrez?db=Genome)
- On the homepage of the NCBI Genome website, it gives links to the major subdivisions of the Genome database, which include Eukaryota, Prokaryota (Bacteria and Archaea), and Viruses. Click on ‘Prokaryota’, since Mycobacterium leprae is a bacterium. This will bring up a list of all fully sequenced bacterial genomes, with the corresponding accession numbers. Note that more than one genome (from various strains) may have been sequenced for a particular species.
- Use ‘Find’ in the ‘Edit’ menu of your web browser to search for ‘Mycobacterium leprae’ on the webpage. You should find that the genomes of several different M. leprae strains have been sequenced. One of these is M. leprae TN, which has accession number NC_002677.
The list of sequenced genomes on the NCBI Genomes website is not a definitive list; that is, some sequenced genomes may be missing from this list. If you want to find out whether a particular genome has been sequenced, but you don’t find it NCBI Genomes website’s list, you should search for it by following these steps:
- Go to the NCBI website (www.ncbi.nlm.nih.gov).
- Select ‘Genome’ from the drop-down list above the search box.
- Type the name of the species you are interested in in the search box (eg. “Mycobacterium leprae”[ORGN]). Press ‘Search’.
Note that you could also have found the Mycobacterium leprae genome sequence by searching the NCBI Nucleotide database, as the NCBI Genome database is just a subset of the NCBI Nucleotide database.
How many genomes have been sequenced, or are being sequenced now?¶
On the NCBI Genome website (http://www.ncbi.nlm.nih.gov/sites/entrez?db=Genome), the front page gives a link to a list of all sequenced genomes in the groups Eukaryota, Prokaryota (Bacteria and Archaea) and Viruses. If you click on one of these links (eg. Prokaryota), at the top of the page it will give the number of sequenced genomes in that group (eg. number of sequenced prokaryotic genomes). For example, in this screenshot (from January 2011), we see that there were 1409 complete prokaryotic genomes (94 archaeal, 1315 bacterial):

Another useful website that lists genome sequencing projects is the Genomes OnLine Database (GOLD), which lists genomes that have been completely sequenced, or are currently being sequenced. To find the number of complete or ongoing bacterial sequencing projects, follow these steps:
- Go to the GOLD website (http://genomesonline.org/).
- Click on the yellow ‘Enter GOLD’ button in the centre of the webpage. On the subsequent page, it will give the number of ongoing bacterial, archaeal and eukaryotic genome sequencing projects.
- Click on the ‘Bacterial Ongoing’ link to see the list of ongoing bacterial genome sequencing projects. By default, just the first 100 projects are listed, and the rest are listed on subsequent pages. In one of the columns of the page, this gives the university or institute that the genome was sequenced in. Other columns give the taxonomic information for the organism, and links to the sequence data.
- Find the number of published genome sequencing projects. Go back one page, to the page with the ‘Bacterial Ongoing’ link. You will see that this page also lists the number of complete published genomes. To see a list of these genomes, click on ‘Complete Published’. This will bring up a page that gives the number of published genomes at the top of the page. In one column of the page, this gives the university or institute that the genome was sequenced in.
As explained above, it is possible to identify genome sequence data in the NCBI Genome database. The GOLD database also gives some information about ongoing genome projects. Often, the GOLD database lists some ongoing projects that are not yet present in the NCBI Genome Database, because the sequence data has not yet been submitted to the NCBI Database. If you are interested in finding out how many genomes have been sequenced or are currently being sequenced for a particular species (eg. Mycobacterium leprae), it is a good idea to look at both the NCBI Genome database and at GOLD.
Summary¶
In this chapter, you have learnt how to retrieve sequences from the NCBI Sequence database, as well as to find out how many genomes have been sequenced or are currently being sequenced for a particular species.
Links and Further Reading¶
There is detailed information on how to search the NCBI database on the NCBI Help website at http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=helpentrez?part=EntrezHelp.
There is more information about the GOLD database in the paper describing GOLD by Liolios et al, which is available at http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2808860/?tool=pubmed.
For more in-depth information and more examples on using the SeqinR package for sequence analysis, look at the SeqinR documentation, http://pbil.univ-lyon1.fr/software/seqinr/doc.php?lang=eng.
There is also a very nice chapter on “Analyzing Sequences”, which includes examples of using SeqinR for sequence analysis, in the book Applied statistics for bioinformatics using R by Krijnen (available online at cran.r-project.org/doc/contrib/Krijnen-IntroBioInfStatistics.pdf).
Acknowledgements¶
Thank you to Noel O’Boyle for helping in using Sphinx, http://sphinx.pocoo.org, to create this document, and github, https://github.com/, to store different versions of the document as I was writing it, and readthedocs, http://readthedocs.org/, to build and distribute this document.
Thank you to Andrew Lloyd and David Lynn, who generously shared their practical on sequence databases with me, which inspired many of the examples in this practical.
Thank you to Jean Lobry and Simon Penel for helpful advice on using the SeqinR package.
Contact¶
I will be grateful if you will send me (Avril Coghlan) corrections or suggestions for improvements to my email address alc@sanger.ac.uk
License¶
The content in this book is licensed under a Creative Commons Attribution 3.0 License.
Exercises¶
Answer the following questions. For each question, please record your answer, and what you did/typed to get this answer.
Model answers to the exercises are given in Answers to the exercises on Sequence Databases.
- Q1. What information about the rabies virus sequence (NCBI accession NC_001542) can you obtain from its annotations in the NCBI Sequence Database?
- What does it say in the DEFINITION and ORGANISM fields of its NCBI record? Note: rabies virus is the virus responsible for rabies, which is classified by the WHO as a neglected tropical disease.
- Q2. How many nucleotide sequences are there from the bacterium Chlamydia trachomatis in the NCBI Sequence Database?
- Note: the bacterium Chlamydia trachomatis is responsible for causing trachoma, which is classified by the WHO as a neglected tropical disease.
Q3. How many nucleotide sequences are there from the bacterium Chlamydia trachomatis in the RefSeq part of the NCBI Sequence Database?
Q4. How many nucleotide sequences were submitted to NCBI by Matthew Berriman?
- Q5. How many nucleotide sequences from nematode worms are there in the RefSeq Database?
- Note that several parasitic nematode worms cause neglected tropical diseases, including Brugia malayi and Wucheria bancrofti, which cause lymphatic filariasis; Loa loa, which causes subcutaneous filariasis; Onchocerca volvulus, which causes onchocerciasis; and Necator americanus, which causes soil-transmitted helminthiasis.
Q6. How many nucleotide sequences for collagen genes from nematode worms are there in the NCBI Database?
Q7. How many mRNA sequences for collagen genes from nematode worms are there in the NCBI Database?
Q8. How many protein sequences for collagen proteins from nematode worms are there in the NCBI database?
- Q9. What is the accession number for the Trypanosoma cruzi genome in NCBI?
- Do you see genome sequences for more than one strain of Trypanosoma cruzi? Note that the Trypanosoma cruzi causes Chagas disease, which is classified as a neglected tropical disease by the WHO.
Q10. How many fully sequenced nematode worm species are represented in the NCBI Genome database?